CFR 21 Part 11, or the FDA’s Electronic Records and Signatures rule, is one of the most important regulations for companies in the life sciences industry. The regulation sets forth specific requirements for companies that use electronic records and signatures in their compliance efforts. This standard is essential for anyone using digital systems to manage data related to products regulated by the FDA. In this blog post, we will provide an overview of CFR 21 Part 11, including a definition of key terms, compliance tips, and information on available tools and certifications.
First of all, let us understand what CFR is…
CFR is the Code of Federal Regulations. CFR 21 Part 11 was published in 1997 and became effective in August of that year. CFR Part 11 applies to all electronic records and signatures created, modified, maintained, archived, retrieved, or transmitted under FDA jurisdiction. This includes records such as laboratory results, audit trails, and software source code listings. It establishes the criteria under which electronic records and signatures are considered trustworthy, reliable, and generally equivalent to paper records.
What is CFR 21 Part 11?
CFR 21 Part 11, often referred to simply as Part 11, is a regulation issued by the U.S. Food and Drug Administration (FDA) that sets forth the criteria for electronic records and electronic signatures in the context of pharmaceuticals, biotechnology, and medical device industries. It is officially titled “Electronic Records; Electronic Signatures” and is part of Title 21 of the Code of Federal Regulations (CFR), which contains FDA regulations.
Here are some key points about CFR 21 Part 11:
- Scope: Part 11 applies to records that are required to be maintained by FDA regulations and which are in electronic format, as well as electronic signatures used to sign such records.
- Purpose: The regulation was introduced to ensure the security, integrity, and reliability of electronic records and signatures in FDA-regulated industries. It helps maintain data accuracy and prevent fraud.
- Requirements: Part 11 outlines specific requirements for electronic records, such as access controls, data security, audit trails, and the use of digital signatures. It also specifies criteria for electronic signatures, including authentication and non-repudiation.
- Applicability: Part 11 is primarily relevant to industries where electronic records and signatures play a critical role in regulatory compliance, including pharmaceutical manufacturing, clinical trials, and laboratory data management.
- Compliance: Companies subject to FDA regulations must comply with the requirements of Part 11 when they use electronic systems to create, modify, maintain, archive, retrieve, or transmit electronic records.
- Validation: One important aspect of compliance with Part 11 is system validation. Firms are expected to validate their electronic systems to ensure they meet the regulatory requirements and function as intended.
- Audit Trails: Part 11 requires the implementation of secure, computer-generated, time-stamped audit trails to record actions that create, modify, or delete electronic records.
- Digital Signatures: Electronic signatures must be linked to their respective electronic records and should be verifiable.
- Record Retention: The regulation also specifies the requirements for the retention of electronic records and the ability to reproduce them in human-readable form when needed.
Compliance with CFR 21 Part 11 is crucial for companies in FDA-regulated industries to ensure the integrity and authenticity of electronic records and signatures. Failure to comply with Part 11 can result in regulatory actions and penalties. Therefore, organizations subject to these regulations invest in systems and processes that meet the requirements laid out in Part 11 to ensure compliance. It’s worth noting that the specific interpretation and implementation of Part 11 requirements can vary between organizations and may require consultation with legal and regulatory experts.
So what does this mean for you and your organization?
If your company uses electronic systems to manage FDA-regulated data, then you are required to comply with CFR 21 Part 11. The regulation sets forth specific requirements for electronic records and signatures, including the use of digital signatures. In order to comply with the standard, you will need to put in place appropriate controls and procedures, as well as create and maintain accurate records. You will also need to provide training for all employees who use electronic systems in their work.
There are a number of tools and services that can help you with CFR 21 Part 11 compliance. For example, EASEUS provides software that automates Part 11 compliance tasks, such as record keeping and signature verification. Validation Master Plan offers Part 11 consulting services to help companies assess their compliance risks and put in place appropriate controls.
CFR 21 Part 11 compliance is essential for any company using electronic systems to manage FDA-regulated data. By putting in place the right controls and procedures, as well as investing in compliance tools and services, you can ensure that your company is compliant with this important regulation.
Critical Components of CFR 21 Part 11:
CFR 21 Part 11, which governs electronic records and electronic signatures in FDA-regulated industries, contains several critical components that organizations must address to ensure compliance. These components help maintain the integrity, authenticity, and security of electronic records and signatures. Here are some of the key components of CFR 21 Part 11:
- Validation: Before implementing electronic systems for regulated activities, organizations must validate them to ensure they meet the intended purposes accurately and reliably. Validation includes verifying that the systems are capable of capturing and maintaining data in a compliant manner.
- Access Controls: Part 11 requires robust access controls to restrict access to electronic records and signatures to authorized personnel only. This involves the use of user authentication mechanisms such as usernames and passwords.
- Data Security: Organizations must implement measures to protect electronic records from unauthorized access, alteration, or deletion. This includes encryption, firewalls, and other security measures to safeguard data.
- Audit Trails: Electronic systems subject to Part 11 must have secure, computer-generated, time-stamped audit trails that record actions taken by users, including the creation, modification, and deletion of electronic records. These audit trails should be readily available for review.
- Electronic Signatures: Part 11 defines requirements for electronic signatures, including methods for creating, storing, and verifying them. Electronic signatures must be linked to their respective electronic records, and there must be controls to prevent unauthorized use of electronic signatures.
- Record Retention: The regulation specifies the requirements for retaining electronic records, including the duration and the ability to reproduce records in human-readable form when necessary. Records must be stored in a manner that ensures their integrity and accessibility.
- System Documentation: Organizations must maintain comprehensive documentation for their electronic systems, including system specifications, procedures, and validation documentation. This documentation is critical for demonstrating compliance.
- Electronic Copies of Records: Part 11 allows for the use of electronic copies of paper records under certain conditions, provided that the copies meet specific requirements related to accuracy, integrity, and availability.
- Electronic Record Amendments: When electronic records are amended or changed, there must be a clear and documented process for making these changes, including an audit trail of the changes and the reasons for them.
- Training and Personnel Qualifications: Organizations must ensure that personnel using electronic systems and electronic signatures are trained and qualified to perform their tasks effectively and in compliance with Part 11.
- Quality Assurance Measures: Establishing a quality assurance program is essential to ensure ongoing compliance with Part 11 requirements. This includes periodic reviews, assessments, and audits of electronic systems and processes.
- Electronic Submission to FDA: For organizations that submit electronic records and data to the FDA, Part 11 outlines requirements for the submission format and procedures to ensure data integrity and security during the submission process.
These critical components of CFR 21 Part 11 are designed to help organizations in FDA-regulated industries maintain the integrity and authenticity of electronic records and signatures. Achieving compliance with these requirements is essential to avoid regulatory issues and penalties and to ensure the quality and reliability of data in the regulated environment.
CFR 21 Part 11 Compliance Tools and Services:
CFR 21 Part 11 compliance is not optional – it is required for all companies that use electronic systems to manage FDA-regulated data. In order to comply with CFR 21 Part 11, you will need to put in place appropriate controls and procedures, as well as create and maintain accurate records. You will also need to provide training for all employees who use electronic systems in their work. There are a number of CFR 21 Part 11 compliance tools and services available to help you with this process.
Achieving and maintaining compliance with CFR 21 Part 11 can be a complex task, and many organizations turn to various tools and services to help them meet the regulatory requirements effectively. These tools and services can aid in electronic record management, electronic signature implementation, data security, validation, and overall compliance efforts. Here are some common tools and services that organizations may utilize for CFR 21 Part 11 compliance:
- Electronic Document Management Systems (EDMS): EDMS software is designed to manage electronic records, ensuring their security, accessibility, and auditability. EDMS systems often include features like electronic signatures, access controls, and audit trails, which can help with Part 11 compliance.
- Electronic Signature Solutions: Electronic signature software provides the capability to create, manage, and verify electronic signatures in compliance with Part 11 requirements. These solutions often integrate with other electronic record management systems.
- Audit Trail Software: Specialized audit trail software can help organizations create and maintain compliant audit trails for electronic records, tracking all actions taken on the records by authorized users.
- Validation Software: Validation software helps organizations validate their electronic systems to ensure they meet Part 11 requirements. It can assist in documenting the validation process and ensuring ongoing compliance.
- Compliance Management Software: These tools are designed to help organizations manage and track compliance efforts across various aspects of Part 11, including documentation, training, and audit activities.
- Secure Data Storage and Backup Solutions: Data storage and backup solutions with robust security features help ensure the integrity and availability of electronic records. These solutions often include encryption and redundancy measures.
- Training and Certification Services: Training providers and consulting firms offer Part 11 compliance training and certification programs for personnel involved in regulated activities. Training is crucial to ensuring that employees understand and follow compliance requirements.
- Regulatory Consulting Services: Regulatory consultants and experts can provide guidance on interpreting Part 11 requirements, assist with validation efforts, and conduct compliance assessments.
- Electronic Submission Services: For organizations that need to submit electronic records and data to the FDA, there are services that specialize in formatting and ensuring the secure transmission of data to meet regulatory standards.
- Document Control and Workflow Automation Tools: These tools help organizations manage document control processes, ensuring that electronic records are reviewed, approved, and distributed in compliance with Part 11.
- Secure Email and Communication Solutions: Organizations often need secure communication channels to exchange electronic records and documents. Secure email and messaging solutions can help meet encryption and access control requirements.
- Cloud Services with Compliance Features: Some cloud service providers offer specialized solutions designed for regulatory compliance, including Part 11. These services often include features for secure data storage and access control.
When selecting tools and services for CFR 21 Part 11 compliance, organizations should consider their specific needs, the complexity of their electronic record systems, and their budget. Additionally, it’s important to ensure that any tools or services chosen align with the organization’s overall compliance strategy and that they meet the specific regulatory requirements outlined in Part 11. Consulting with regulatory experts or legal counsel may also be beneficial in navigating the compliance landscape.
Certifications Available for CFR 21 Part 11 Compliance:
There are a number of certifications available for CFR 21 Part 11 compliance. These include the Certified CFR 21 Part 11 Professional (CCP) certification from the Regulatory Affairs Professionals Society (RAPS), as well as the Certified Quality Systems Auditor (CQSA) certification from ASQ.
The CCP certification is designed for professionals who work with CFR 21 Part 11 compliance on a daily basis. The certification covers topics such as CFR 21 Part 11 requirements, records and signatures, compliance risks, and mitigation strategies.
The CQSA certification is for quality professionals who want to demonstrate their knowledge of quality systems auditing. The certification covers topics such as quality principles, audit planning, conducting an audit, and reporting results.
CFR 21 Part 11 compliance is essential for regulated companies using electronic records and signatures. There are a number of certifications available that can help you demonstrate your knowledge and understanding of the regulation.
By investing in Part 11 compliance tools and services, as well as pursuing certifications, you can ensure that your company is compliant with this important regulation.
Prerequisites for CFR 21 Part 11:
There are 7 critical requirements for CFR 21 Part 11 compliance.
1. Data integrity
The EPA’s Part 11 rules state that you must have a digital process and controls in place to guarantee the “authenticity, integrity, and, when appropriate, confidentiality of electronic records.”
The main goal of Part 11 is to make sure the data and information you gather while developing your product are accurate, traceable, suitable for your purpose, and secure from loss or misuse. The risk of product failure will be reduced by imposing all of Part 11’s safeguards. It’s a smart investment.
2. Data retrieval
The regulations state that you should have the tools to preserve your documentation “to enable their timely and accurate retrieval throughout the records retention period.” Controlling the development process so that your records are automatically archived, indexed, and accessible on-demand will assist you in:
- Examining and checking for non-compliance and problems, audit your own system thoroughly.
- Tracking and tracing ‘the root causes of any identified non-conformities in your system
- Supporting external audits – respond quickly to regulatory questions to keep your business compliant
3. Validation
You must formally describe how your system is supposed to function, then write scripts and testing procedures to ensure it functions as intended. However, it may seem like a lot of work, but validating your QMS will show that it is fit for purpose and give you and the regulator assurance that you are capable of delivering goods according to the required standards.
4. Audit Trails
Part 11 requires you to have a complete version history available for every quality document in your system, via secured, computer-generated, and time-stamped audit trails in order to independently record the date and time of operation entries and actions that create, modify, or delete electronic records.
The more details you can get on every change and sign-off event, the better. By recording the author, date, and time of each modification and sign-off event, you’ll have full traceability and accountability for all decision-making activities throughout the development process. It will save time and resources than using a paper-based system in auditing and investigating procedures.
5. Operational Controls
The usage of operational system checks so as for the enforcement of appropriate sequential steps and events is another important requirement of Part 11.
The ability to create automated workflows for obtaining approvals and signatures will give you more control over your team and procedure as you manage the implementation process. They can ensure important papers are collected together before being examined by various people at specific times in your plan. Part 11 aids in the creation of order and clarity in potentially complicated procedures, lowering the danger of a company making costly errors.
6. Security Controls
Part 11 sets out the controls you’ll have over who has access and how it’s changed within your system. The rules include several precise criteria to prevent data loss and deletion by accident, as well as security breaches that can cause customer damage, business failure, and government fines.
7. Electronic signatures
Part 11 mentions the mandatory usage of e-signatures.
Part 11 requires that electronic signatures be digitally signed documents with a printed name of the signer, the date/time the signature was applied, and the ‘meaning’ or purpose of the electronic signature as part of an evolving and uneditable audit trail. However, this is not where things end.
In an effort to match the level of legal confidence provided by a ‘wet signature,’ Part 11 has made digital approval authentication procedures far more demanding. To guarantee identity verification and protection from fraud, you’ll need tight controls over digital documents and processes. It would be significantly simpler to fabricate a pen and ink signature on a test result than it is to falsify an electronic signature under FDA rules right now.
Visure Requirements ALM Platform:
Visure is one of the most trusted modern ALM platforms that specializes in requirements management for organizations of all sizes across the globe. It’s a must-have tool for teams building complex products, systems, and software, which require end-to-end traceability from conception to testing and deployment, all the way to source code, along with standard certification compliance.
The CFR 21 Part 11 Module in Visure Requirements ALM Platform is a complete solution for companies who need to comply with CFR 21 Part 11. The module includes a CFR 21 Part 11 checklist, which helps you assess your company’s compliance status, and a CFR 21 Part 11 template, which can be used to create CFR 21 Part 11 compliant quality documents.
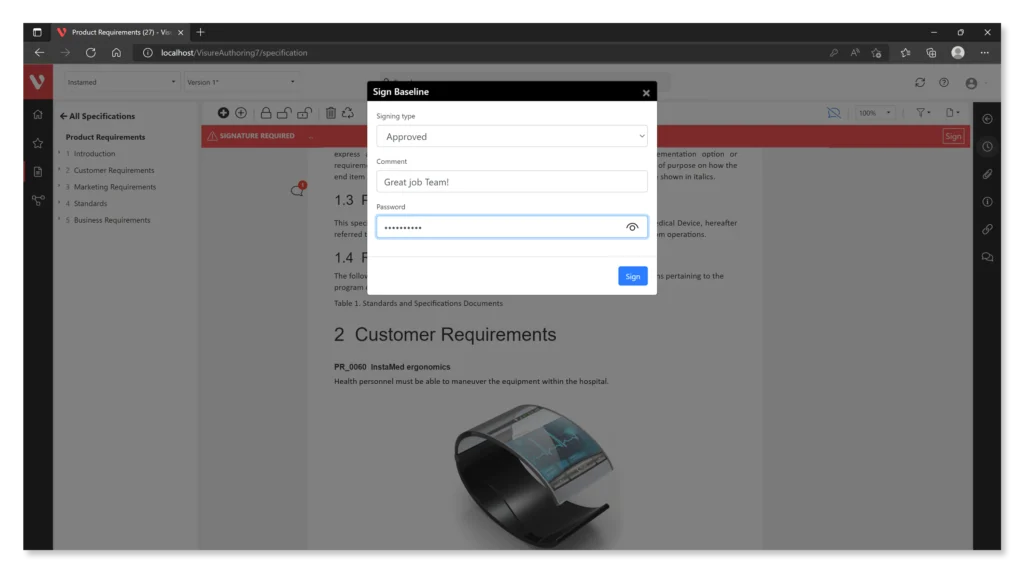
Here are some features offered by Visure:
- Electronic Signature Support: The software should allow for the creation and management of compliant electronic signatures, ensuring that they are secure, verifiable, and linked to the associated electronic records.
- Audit Trail Functionality: Robust audit trail capabilities enable organizations to maintain secure, time-stamped records of all actions taken on electronic records, including creation, modification, and access.
- Access Controls: The software should provide access control features to restrict access to electronic records and ensure that only authorized personnel can view, edit, or approve them.
- Data Security: Strong data security measures, such as encryption and user authentication, are essential to protect electronic records from unauthorized access or tampering.
- Electronic Record Storage and Retrieval: The system should facilitate the secure storage and retrieval of electronic records, ensuring their integrity and availability as needed.
- Validation and Compliance Tools: Compliance software often includes tools to help organizations validate their systems and processes to ensure they meet Part 11 requirements. This may include validation documentation templates and workflows.
- Documentation and Reporting: The software should assist with the creation and management of documentation required for compliance, such as user manuals, standard operating procedures (SOPs), and compliance reports.
- Training and User Management: Tools for managing user access and training records can help ensure that personnel are appropriately trained and authorized to use the system in compliance with Part 11.
Conclusion:
CFR 21 Part 11 is a set of regulations from the United States Food and Drug Administration (FDA) that apply to electronic records and signatures in the life sciences industry. The key components of CFR 21 Part 11 are authentication, integrity, security, and retention. To comply with the standard, companies must have a system for tracking changes to electronic records, authenticating user actions, and maintaining audit trails. Visure Requirements ALM Platform helps companies meet these requirements by providing a secure platform for managing requirements and change management processes. Request a free 30-day trial at Visure Requirements ALM Platform today to see how our software can help you achieve compliance.